
REC articles are not the view or opinion of Alpha Extract Administrators
Hemorrhage in the Brain Causes Damage that Cannabinoids Can Stop
Omega8 is well known throughout the cannbis community as "Miraculous"

Omega8 truly miraculous | alphaExtract (alphaextracts.ca)
Hemorrhage in the Brain Causes Damage that Cannabinoids Can Stop
CB2 receptors are activated following hemorrhage on the brain, and this is where cannabinoids will help stop further damage.
When the brain suffers a stroke or a traumatic injury, damage to the brain continues as the hemorrhage leads to a snowball effect of inflammation, oxidative stress, and excitotoxicity, or cell death by over-stimulation, usually by the neurotransmitter glutamate.
- Advertisement -
There aren’t many medications that can be given immediately following brain injury or hemorrhage to mitigate these effects, but because the endocannabinoid system (ECS) is involved in all of these mechanisms, cannabinoids could be the perfect treatment.
The activity of the ECS has been studied extensively in animal models of stroke and traumatic brain injury (TBI). These studies have found that the expression of endocannabinoids and cannabinoid receptors in the brain is unregulated following stroke or TBI. For example, levels of the endocannabinoid 2-arachidonoylglycerol (2-AG) spiked following an injury and remained elevated for more than 24 hours. This indicates that the ECS could be working to protect the brain in response to injury. In fact, 2-AG acts as a vasorelaxer, so it improves blood flow in the brain after injury.
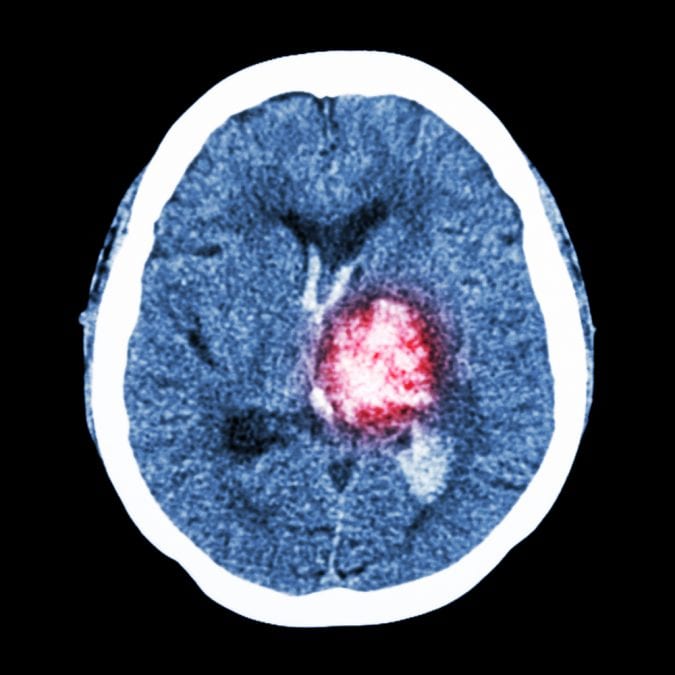
Many animal studies looked at the role of cannabinoid receptor CB2 in protecting the brain from damage following TBI or stroke. When cannabinoids that activate the CB2 receptor (also called receptor agonists) were administered prior to the simulation of a stroke , th damaged and dead tissue was much smaller than in animals that did not receive the treatment. Further confirmation of the role of CB2 was found after administering a cannabinoid that reduces or blocks the activity of the CB2 receptor; the infarct size increased.
Activating the CB2 receptor seems to primarily work through the suppression of the inflammatory response. CB2 receptor agonists reduce the ability of white blood cells to accumulate in the injury site during a stroke by limiting their ability to stick to the injured tissue. The reduction of expression of intracellular adhesion molecule (ICAM-1) at the injury site as a result of CB2 activation is likely the mechanism responsible for this. When the CB2 gene was deleted in mice, then ICAM-1 expression increased, as did pro-inflammatory cytokine tumor necrosis factor alpha (TNF-α). Inhibiting the release of cytokines like TNF-α is one of the known anti-inflammatory mechanisms of CB2 activation. The reduction of white blood cell recruitment to the injury site also protects the blood-brain barrier, which helps to shield the injury site from worsening inflammation.

The CB2 receptors are also highly expressed on microglial cells, a kind of brain cell that is actually related to macrophages and migrates to the injury site. Microglia contribute to the inflammation response that can cause death of neurons in the area. However, activating the CB2 receptors seems to reduce the effect the cells have, preventing their release of toxic compounds and pro-inflammatory cytokines.
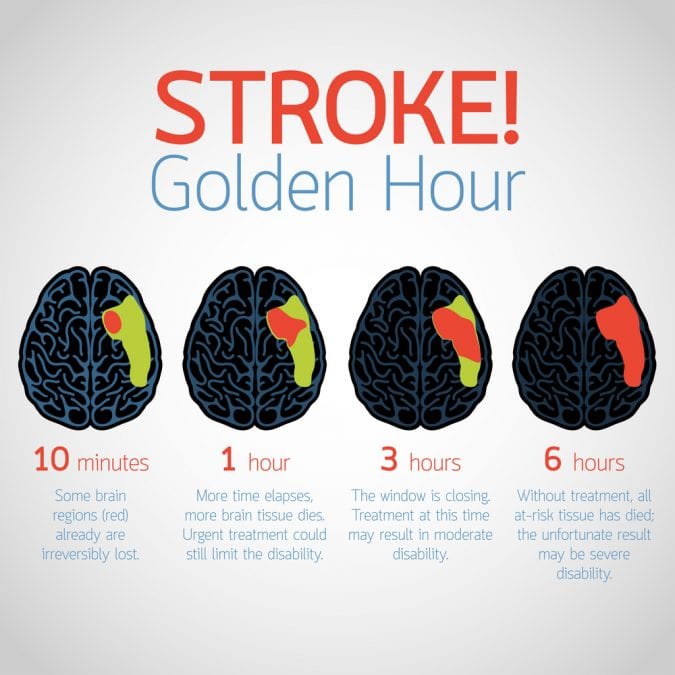
The CB1 receptor may also play an important role in protecting the brain from excitotoxicity. The neurotransmitter glutamate is released in large amounts following brain injury, which leads to cell death. However, CB1 activation inhibits various pathways involved in glutamate signaling, preventing glutamate release. The CB1 receptor may also be involved in antioxidant pathways, since 2-AG was found to reduce the release of reactive oxygen species (ROS) following injury. Cannabinoids themselves are also known to be effective antioxidants, and so may work through other pathways to reduce the effects of ROS release at the injury site.
- Advertisement -
The large number of studies on the role of cannabinoids and the ECS in mitigating the effects of stroke or TBI are very encouraging. However, there are no human clinical trials that have been performed to date to demonstrate the efficacy of cannabinoids at reducing the brain damage following TBI or stroke.
Omega8 Copied By Many But Never Duplicated !!!

In order to Post a comment - Create an account or Sign in
Related Articles